As Glicogenoses ou Doença do Depósito de Glicogênio (Glicogen Storage Disease – GSD) são doenças de causa genética, que resultam em alterações no metabolismo do glicogênio. As glicogenoses são consideradas doenças raras, considerando a frequência estimada global de 1/20.000 a 1/25.000 nascidos vivos.
Como é viver com Glicogenose
A primeira grande dificuldade já se encontra no diagnóstico, que pode demorar meses. Por se tratar de uma doença rara, há dificuldade de encontrar profissionais de saúde que realmente conheçam sobre ela e principalmente sobre seu controle. Não há remédios para tratá-la e nem cura, mas com conhecimento e controle rigoroso e adequado, é possível ter um desenvolvimento praticamente normal, permitir uma melhor qualidade de vida para os pacientes e a ausência das várias sequelas que podem ser graves e fatais. Daí a importância de se conhecer sobre a doença!
A glicogenose é um erro inato do metabolismo, uma doença hereditária na qual a deficiência de uma enzima afeta o processamento da síntese do glicogênio, ou a sua quebra em glicose nos músculos e fígado, resultando no depósito de glicogênio e em falta de energia para o organismo, causando hipoglicemias graves e frequentes. Desse modo, os pacientes precisam se submeter a uma restrição alimentar severa, não podendo consumir muitos alimentos, como lactose, frutose e sacarose. Além da restrição, para manutenção da glicemia é necessário o uso constante de amido de milho cru, dia e noite, que varia de acordo com idade, peso e necessidade metabólica de cada pessoa.
Viver com a glicogenose é viver em um turbilhão de emoções. Da incerteza do diagnóstico, do medo de se perder um filho ou de viver com a doença,
das incertezas do desenvolvimento, da falta de conhecimento e orientação, dos riscos diários, dos altos custos, do cansaço físico e mental.
Mas por fim, o que prevalece é o amor, a garra e a força para vencer esse grande desafio.
Tipos de Glicogenose
As glicogenoses podem ser classificadas em diferentes tipos, de acordo com o defeito enzimático específico e os órgãos afetados. É muito importante saber qual o tipo de glicogenose cada pessoa tem, pois cada tipo é causado por diferentes genes e diferentes enzimas, que acarretam em variações nas manifestações clínicas e nas formas de tratar as doenças.
Os tipos mais comuns são os I, II, III e IX. No Brasil não há um levantamento preciso da incidência de cada tipo, mas acredita-se que os tipos mais comuns sejam os tipos I e III (dados obtidos de levantamento dos pacientes do Hospital de Clínicas de Porto Alegre e da Universidade de Campinas).
Hipoglicemia, hepatomegalia, retardo do crescimento, acidose lática, hiperuricemia e hiperlipidemia.
Acompanhado de neutropenia (diminuição do número de neutrófilos no hemograma); infecções bacterianas recorrentes;
pode ocorrer doença inflamatória intestinal.
Gene:
SLC37A4
Tratamento:
Uso de amido de milho cru; restrição de galactose, frutose, lactose e sacarose.
Uso de estimulador de colônia de neutrófilos e tratamento da doença inflamatória intestinal.
Pode ter apresentação infantil, com hipotonia, dificuldade de sucção e cardiomiopatia;
ou apresentação adulta com doença muscular progressiva, cardiomiopatia e dificuldades respiratórias.
Gene:
GAA
Tratamento:
Fisioterapia motora e respiratória, terapia de reposição enzimática.
Hepatomegalia, hipoglicemia cetótica, retardo do crescimento, hiperlipidemia, elevação da AST e ALT, CPK.
Fraqueza muscular e cardiomiopatia ocorrem no subtipo IIIa.
Gene:
AGL
Tratamento:
Uso de amido de milho cru; dieta hiperproteica; restrição de sacarose.
Miopatia, contraturas articulares, epilepsia, atraso de desenvolvimento, catarata.
Na forma adulta, pode se apresentar com fraqueza muscular progressiva.
Gene:
PFKM
Tratamento:
Dieta rica em proteínas e vitamina B6; evitar exercício extenuante.
Hepatomegalia, hipoglicemia cetótica ao jejum, retardo do crescimento, elevação de AST/ALT, hiperlipidemia leves e cetonas aumentadas no sangue e urina.
Podem haver sintomas musculares e elevação da enzima muscular CpK.
Gene:
PHKA2
Tratamento:
Uso de amido de milho cru; dieta rica em proteínas; evitar grandes quantidades de sacarose.
Hepatomegalia, hipoglicemia cetótica ao jejum, retardo do crescimento, elevação de AST/ALT e hiperlipidemia leves.
Cirrose hepática pode se desenvolver.
Gene:
PHKG2
Tratamento:
Uso de amido de milho cru; dieta rica em proteínas; evitar grandes quantidades de sacarose.
Hipoglicemia, déficit de crescimento, raquitismo e abdome protuberante devido ao aumento do tamanho do fígado e rins.
Gene:
GLUT2
Tratamento:
Restrição da ingestão de galactose, suplementação de água, amido de milho, eletrólitos e vitamina D.
A Glicogenose Ia é também conhecida como Doença de Von Gierke, e possui um padrão de herança autossômico recessivo. Caracteriza-se pela deficiência da atividade da enzima glicose-6-fosfatase (G6Pase), que se localiza no retículo endoplasmático sendo expressa principalmente no fígado, rins e intestino. O gene G6Pase localiza-se no cromossomo 17q21.31. Já a Glicogenose Ib ocorre por uma alteração no transportador de glicose-6-fosfato (G6PT), e o gene SLC37A4 foi mapeado no cromossomo 11q23.3. A GSD Ib representa cerca de 20% de todos os casos de GSD I, sendo menos comum do que a GSD Ia.
DIAGNÓSTICO: As GSD tipo I tem incidência estimada de 1:100.000, correspondendo a 25% de todos os tipos de glicogenose. O diagnóstico da maioria dos pacientes com GSD I pode ser realizado pela combinação de achados clínicos, testes bioquímicos e genéticos. Pode ser realizado o estudo do sistema G6Pase que requer uma amostra hepática de fígado não congelado. A medida da atividade de G6Pase é realizada em microssomas intactos e em microssomas rompidos. A GSD Ia é caracterizada pela deficiência de atividade de G6Pase em microssomas hepáticos intactos e rompidos, já na GSD Ib a atividade de G6Pase é deficiente em microssomas intactos e (sub) normal em microssomas rompidos. Na maioria dos pacientes com GSD Ia, a atividade residual da G6Pase é inferior a 10% do normal. O diagnóstico da GSD Ia pode ser ainda estabelecido pela análise do gene G6PC. Em pacientes que possuem as características clínicas e bioquímicas de GSD I, associadas a neutropenia e/ou infecções recorrentes, deve ser feita a pesquisa de mutações em G6PT (achados comuns em GSD Ib).
ALTERAÇÕES METABÓLICAS significativas ocorrem nesses pacientes, resultado da produção endógena de glicose alterada ou ausente e pelas rotas alternativas que a glicose-6-fostato segue, como: hipoglicemia, aumento de lactato, aumento de ácido úrico, hipertrigliceridemia e hiperlipidemia, especialmente após um curto período de permanência em jejum.
SINTOMAS podem ser observados ao nascimento ou nas primeiras semanas de vida. O mais comum é o aparecimento, aos 3 – 4 meses de vida, de hepatomegalia ou convulsão associada à hipoglicemia. Os bebês costumam apresentar hipoglicemia após pequenos períodos de jejum ou após infecções. A hipoglicemia se caracteriza por palidez, suor frio, irritação, convulsões. Alterações frequentemente descritas incluem face de boneca, obesidade troncular, abdômen globoso pela hepatomegalia, resultante do acúmulo de glicogênio e esteatose hepática, hipotonia e baixa estatura. Também são descritos anemia, manifestações intestinais, como diarreia intermitente, enteropatia relacionada à GSD e dano da mucosa intestinal. A GSD I também se manifesta com doença renal progressiva, alterações hepáticas teciduais tais como hepatócitos edemaciados, esteatose, hiperglicogenação nuclear, e adenomas hepáticos em 22%-75% dos pacientes adultos. Os adenomas hepáticos podem ser solitários ou múltiplos e como complicação podem apresentar hemorragia ou malignização (risco de 10%). A regressão dos adenomas com a instituição da dieta adequada é descrita na literatura.
Com tratamento inadequado, a acidose metabólica crônica pode afetar a atividade do hormônio de crescimento, resultando no retardo do crescimento e a baixa estatura. Evidências mostram que um bom controle metabólico desses pacientes pode favorecer o crescimento. Alguns pacientes apresentam osteoporose, a qual pode estar relacionada à nutrição inadequada, à falta de vitaminas, aos efeitos do ácido láctico e ao hipogonadismo. Na GSD Ib é descrita ainda neutropenia crônica e déficit de função de neutrófilos e monócitos. A neutropenia resulta em infecções bacterianas recorrentes, além de úlceras orais e na mucosa intestinal. A doença inflamatória intestinal semelhante à Doença de Crohn é um achado frequente na GSD Ib, sendo usualmente a maior causa de morbidade nesses pacientes.
TRATAMENTO da GSD I é basicamente dietético e tem como objetivo proporcionar uma fonte contínua de glicose, manter normoglicemia, ou seja, níveis de glicose acima de 4mmol/L, ou 70 mg/dL, e prevenir distúrbios metabólicos secundários.
As estratégias atuais de tratamento incluem dieta contínua noturna por sonda nasogástrica, gastrostomia e/ou administração frequente e a intervalos regulares de amido de milho cru (AMC), na dose de 1,75 a 2,5g/kg de peso a cada 4 a 6 horas, com uso de balança. Por ser um polissacarídeo, o AMC apresenta uma degradação lenta e mantém assim a normoglicemia plasmática. A dose de AMC para os pacientes com GSD I varia de acordo com a idade e o controle metabólico do paciente, com intervalos de 3-5 horas durante o dia e 4-6 horas no período da noite. Em bebês pode ser necessária a manutenção de uma dieta contínua, ou redução do intervalo de administração das dietas, de acordo com a necessidade metabólica a fim de manter a glicemia dentro da normalidade.
A distribuição calórica da dieta dos pacientes deve ser 60 a 65% de carboidratos, sendo destes, 30 a 45% sob a forma de amido de milho cru, 20 a 25% de lipídeos e 10 a 15% de proteínas. A SACAROSE, FRUTOSE, LACTOSE e GALACTOSE devem ser rigidamente restritas, pois transformam-se em glicose. São absorvidas rapidamente e transformam-se em glicogênio, aumentando progressivamente o glicogênio hepático, gerando uma extensa lista de alimentos que não são permitidos na dieta desses pacientes.
A glicemia deve ser monitorada DIARIAMENTE, possibilitando a realização de ajustes no uso do amido de milho, tanto para dose quanto para horários, além de ajustes na prescrição da dieta.
As suplementações de vitaminas e minerais devem ser realizadas de acordo com o tipo de dieta oferecida e a idade, de acordo com as recomendações da Organização Mundial de Saúde. As restrições dietéticas, como de frutas e produtos lácteos, e que focam na manutenção da normoglicemia, podem resultar em limitada oferta e deficiências nutricionais por exemplo, de cálcio, ferro e vitamina D. Os níveis subótimos de vitaminas possivelmente são causadas pela natureza restritiva da dieta, as alterações metabólicas e a má absorção intestinal.
O tratamento farmacológico da GSD I objetiva principalmente os controles da hiperuricemia (o alopurinol pode ser usado nos casos em que o manejo dietético não for suficiente para controlar os níveis de ácido úrico), da acidose persistente e da perda urinária de proteína. Para GSD Ib, acrescenta-se ainda o uso de estimulador de colônia de neutrófilos (filgastrima).
O transplante de fígado é uma opção terapêutica apenas para os pacientes que apresentam complicações graves como descompensação metabólica frequente, atraso grave no desenvolvimento e surgimento de neoplasias.
A glicogenose tipo II (Doença de Pompe) é uma doença grave e muitas vezes fatal, causada pela deficiência da enzima alfa-glicosidase ácida, levando a um acúmulo de glicogênio lisossômico nas células dos tecidos musculares. É uma doença neuromuscular que pode progredir de forma variada.
SINTOMAS: início precoce de problemas respiratórios anteriores à fraqueza dos membros. A forma infantil manifesta-se nos primeiros meses de vida, com fraqueza muscular, dificuldade de sucção e cardiomiopatia. Por outro lado, a forma tardia manifesta-se em crianças com idade mais avançada e em adultos com insuficiência, respiratória sendo precedida de fraqueza muscular.
TRATAMENTO: envolve a fisioterapia motora e respiratória e a terapia de reposição enzimática.
A GSD III, também conhecida como Doença de Cori ou Doença de Forbe, é uma doença autossômica recessiva que ocorre quando há deficiência da enzima desramificadora (amilo-1,6-glicosidase). A liberação de glicose do glicogênio requer a ação tanto da enzima glicogênio fosforilase quanto da enzima desramificadora. Quando há ausência ou deficiência da enzima desramificadora, a glicogenólise é interrompida nos pontos de ramificação mais externos. Como consequência, ocorre o acúmulo anormal de glicogênio nos órgãos afetados (fígado, coração, musculatura esquelética, leucócitos no tipo IIIa, e fígado no tipo IIIb). O gene AGL localiza-se no cromossomo 1p21.2.
Existem dois subtipos principais de GSD III: o tipo IIIa, o qual afeta tanto o fígado quanto os músculos, e compreende cerca de 85% dos casos, e o tipo IIIb que afeta somente o fígado e compreende cerca de 15% de todos os pacientes com GSD III. Esses dois fenótipos clinicamente distintos são causados por mutações no mesmo gene (AGL) e são explicados por diferenças na expressão da enzima deficiente em diferentes tecidos. É descrita uma incidência estimada de 1:100.000 para a doença.
DIAGNÓSTICO: O diagnóstico se inicia a partir da suspeita clínica (hepatomegalia, hipoglicemia, alterações de transaminases e creatinaquinase), e pode ser confirmado por meio da medida anormal de glicogênio no fígado/músculo ou por meio da medida da atividade da amilo-1,6-glicosidase no fígado, músculo e nos eritrócitos. A medida da enzima no músculo é necessária para diferenciar os subtipos IIIa e IIIb, pois pacientes com GSD IIIa podem apresentar função muscular normal, bem como níveis normais de creatina quinase na idade adulta, e podem ser erroneamente diagnosticados com GSD IIIb. A análise de mutações é um método diagnóstico fácil e não invasivo e pode ser feita a partir de amostras de sangue, biópsia do fígado, músculo esquelético e cardíaco, eritrócitos e fibroblastos cultivados.
SINTOMAS: Os indivíduos com GSD III apresentam hepatomegalia, hipoglicemia, baixa estatura, miopatia esquelética e miocardiopatia. A maioria dos pacientes tem tipo IIIa e apresentam envolvimento hepático e muscular, já os pacientes com tipo IIIb, apresentam apenas envolvimento hepático. A doença hepática ocorre progressivamente ao longo da vida com o desenvolvimento da fibrose do fígado e, em alguns casos, cirrose e carcinoma hepatocelular. Adenomas hepáticos têm sido observados em 25% dos pacientes com GSD III, contudo, a relação entre a formação de lesões e o controle metabólico ainda não é bem-conhecida. Alguns pacientes apresentam hipertrigliceridemia, mas ácido úrico e lactato costumam estar dentro da faixa considerada normal. A presença de cetose urinária e plasmática é um marcador bioquímico de mau controle metabólico.
Cardiomiopatia hipertrófica ocorre na maioria dos indivíduos com tipo IIIa, mas sua significância clínica continua incerta e a maioria dos pacientes são assintomáticos. A miopatia está ausente ou é mínima na infância e progride lentamente, tornando-se proeminente na terceira ou quarta década de vida. Osteoporose e osteopenia têm sido observadas em GSD III, assim como em outras doenças de depósito de glicogênio, provavelmente associadas à baixa ingestão de cálcio. Além disso, pode ser encontrada a doença do ovário policístico, mas a fertilidade não parece ser afetada. O crescimento desses pacientes pode ser comprometido pelo mau controle metabólico, contudo a retomada do crescimento pode ser observada com o estabelecimento de um bom controle metabólico, baseado em uma dieta adequada, indicada para GSD. Os sintomas podem ser considerados menos graves que na GSD I, sendo que a tolerância ao jejum sofre variação e a hipoglicemia em geral é menos grave.
TRATAMENTO: O principal foco do tratamento é a oferta regular de glicose, a fim de manter seus níveis sanguíneos acima de 70 mg/dL. A terapia com amido de milho cru é prescrita com dose de acordo com a idade e o controle metabólico do paciente a cada 4-6 horas, sendo capaz de manter a normoglicemia, aumentar a velocidade de crescimento e diminuir a concentração das transaminases. Galactose e frutose podem ser utilizadas, contudo as suas quantidades devem ser adaptadas a cada paciente, evitando o armazenamento excessivo de glicogênio.
A gliconeogênese está preservada nesses pacientes, sendo recomendada uma ingestão de 3g/kg/dia de proteína, visto que essa pode ser utilizada como uma fonte de energia eficaz. A alta ingestão proteica impede ainda a quebra de proteína muscular em momentos de jejum, preservando assim os músculos esqueléticos e cardíacos. A dieta recomendada para esses pacientes compreende valores de 55-60% de carboidratos, 15-20% de proteína e 20-25% de lipídeos.
Devido à possibilidade de osteopenia e osteoporose, a suplementação de vitamina D e cálcio também é recomendada nessa condição.
A glicogenose tipo IV (Doença de Andersen) é causada pela deficiência da enzima amilo-1,4-1,6- transglicosidase, essencial na ramificação do glicogênio. O acúmulo de corpúsculos de poliglicosana, principalmente no fígado e no músculo, resultado da atividade residual da enzima, é avaliado para o diagnóstico da doença.
SINTOMAS: As manifestações clínicas relacionadas a essa doença são, principalmente, hepatoesplenomegalia (aumento do fígado e do baço), cirrose, hipotonia muscular, falhas no crescimento, devastação muscular nas extremidades inferiores, disfunção neuromuscular e desenvolvimento motor lento.
TRATAMENTO: Não há tratamento eficaz definido e o transplante de fígado é a única modalidade curativa.
A glicogenose tipo V (doença de McArdle) é causada pela deficiência na enzima miofosforilase, o que impede que o glicogênio seja direcionado para a hidrólise, que resulta no seu acúmulo nos músculos. A condição é autossômica recessiva, causada por mutações no gene PYGM (11q13), levando à deficiência de fosforilase muscular. A mutação P.R50X é responsável por 40% a 50% dos alelos em populações caucasianas. É uma forma grave da doença de armazenamento de glicogênio, caracterizada pela intolerância ao exercício.
DIAGNÓSTICO: baseado na não elevação dos níveis de lactato no sangue durante um teste de exercício físico, pelo excesso de glicogênio, pela deficiência na atividade enzimática em biópsia muscular e por estudos moleculares que avaliam variações no gene da enzima miofosforilase. O diagnóstico diferencial deve incluir a GSD tipo VII.
SINTOMAS: são relacionados à intolerância ao exercício muscular, podendo apresentar mialgia, câimbras, fadiga muscular excessiva e fraqueza muscular. Um alívio da mialgia e da fadiga depois de alguns minutos de descanso é observado em muitos doentes. Em cerca de 50% dos acometidos pela doença, após a realização de exercícios é observada elevação da creatina quinase e rabdomiólise com mioglobinúria (urina escura), podendo levar à insuficiência renal aguda. Após os 40 anos, as câimbras e a mioglobinúria tornam-se menos intensas, e a fraqueza muscular e as amiotrofias ficam mais evidentes. O prognóstico pode ser favorável ao se evitar a rabdomiólise grave, contudo, a mioglobinúria pode levar à insuficiência renal com risco de morte. Foram descritos casos com manifestação precoce de hipotonia, fraqueza muscular generalizada e insuficiência respiratória progressiva.
TRATAMENTO: controle do exercício físico.
A glicogenose tipo VI (Doença de Hers) é causada pela deficiência da enzima fosforilase hepática no fígado, pela mutação no cromossomo 14. A enzima está presente ainda em células musculares e nervosas, porém as alterações são decorrentes de mutações nos cromossomos 11 e 20, respectivamente. A enzima glicogênio fosforilase catalisa, juntamente com a enzima desramificadora, a degradação de glicogênio em glicose-1-fosfato, sendo essencial no armazenamento de glicose na forma de glicogênio.
SINTOMAS: hepatomegalia, fraqueza muscular, hemorragia nasal, crise de hipoglicemia, leve retardo de crescimento, boa tolerância ao jejum, elevação de triglicérides, colesterol e ácido úrico.
TRATAMENTO: dieta semelhante ao tipo III, com pouca necessidade de tratamento. Se sintomático, aumento de carboidrato e alimentação frequente e dieta hiperproteica.
A glicogenose tipo VII (Doença de Tarui) ocorre devido a um defeito na enzima fosfofrutoquinase, que leva ao acúmulo de glicogênio no músculo esquelético, como resultado de um bloqueio na metabolização da glicose-6-fosfato pela via glicolítica.
SINTOMAS: são heterogêneos, como hemólise, morte durante a infância e intolerância ao exercício, devido à fraqueza muscular, fadiga fácil, câimbras pós-exercícios, atraso de desenvolvimento, catarata, epilepsia. A sintomatologia é semelhante à da doença de McArdle, desse modo, o diagnóstico diferencial é possível pela histoquímica da biópsia do músculo.
TRATAMENTO: Dieta rica em proteínas e vitamina B6; evitar exercício extenuante
A glicogenose tipo VIII caracteriza-se por um defeito na função da enzima fosforilase-bquinase, envolvida no metabolismo do glicogênio, a partir da regulação da enzima glicogênio fosforilase. A alteração pode ser originada pelas mutações dos genes PHKA2, PHKB e PHKG2, subunidades da fosforilase quinase.
O diagnóstico preciso é determinado pela análise do tecido hepático ou pelo teste genético.
SINTOMAS: manifestam-se ainda na infância, com hepatomegalia, retardo do crescimento e níveis aumentados de lipídeos e aminotransferase. Com frequência ocorre completa resolução dos sintomas na puberdade. Uma minoria dos pacientes apresenta um fenótipo mais grave, com hipoglicemia de jejum sintomático e histologia hepática anormal que pode progredir para cirrose.
A GSD IX é causada pela deficiência da enzima fosforilase quinase hepática ou muscular, e pode ser dividida em subtipos. A GSD IXa é o subtipo mais comum, sendo causada por mutações em PHKA2, com localização citogenética em Xp22.13 e tem padrão de herança recessivo ligado ao X.
A GSD IXb é causada por alterações no gene PHKB, com localização citogenética em 16q12.1 e tem padrão de herança autossômica recessiva. A GSD IXc é causada por mutações em PHKG2, com localização citogenética em 16p11.2 e tem padrão de herança autossômica recessiva.
O subtipo C acomete também as células sanguíneas. É descrita uma incidência estimada de 1:100.000 para deficiência de PHK hepática.
DIAGNÓSTICO: pode ser realizado em diferentes tecidos, mas sua sensibilidade é baixa pelas diferentes isoformas da enzima que esses apresentam. Análises da atividade da enzima fosforilasequinase possibilitam o diagnóstico laboratorial dessa glicogenose.
SINTOMAS: As manifestações clínicas da deficiência de fosforilase quinase hepática se iniciam nos primeiros anos de vida, sendo característicos hepatomegalia e retardo de crescimento, que é mais pronunciado na infância, e na vida adulta os pacientes podem alcançar estatura normal. Podem ser encontrados também hipoglicemia hipercetótica geralmente leve, a puberdade pode ser atrasada e presença de fibrose hepática que pode progredir mais raramente para cirrose. São descritos ainda elevação de transaminases hepáticas, hipercolesterolemia e hipertrigliceridemia. Já as manifestações clínicas relacionadas à enzima muscular são intolerância ao exercício, mialgia, cãibras musculares, mioglobinúria e fraqueza muscular progressiva
Apesar de ser tipicamente classificada como um tipo leve de GSD, alguns pacientes podem apresentar sintomas típicos de GSD I e III, com hipoglicemia, grave hepatomegalia e aumento de transaminases.
TRATAMENTO: visa a prevenir a hipoglicemia e a cetose. Preconiza-se por dieta frequente e complementada com a utilização de Amido de Milho Cru, com dose e frequência de acordo com a idade, e a tolerância ao jejum e com os sintomas clínicos. O percentual de consumo proteico varia entre 15 e 25%, visto que a via de gliconeogenêse está preservada, essa pode ser uma fonte de glicose. Portanto, recomenda-se de 3-3,5g/kg/dia de proteína.
A glicogenose tipo X é um transtorno causado pela deficiência da enzima fosfoglicerato mutase, que leva ao acúmulo de glicogênio nos músculos. Relevante é a associação comum com agregados tubulares provenientes do retículo sarcoplasmático, visualizadas em biópsias musculares, não presente nas outras glicogenoses.
SINTOMAS: intolerância aos exercícios, câimbras, mioglobinúria e fraqueza.
A glicogenose tipo XI (síndrome de Fanconi-Bickel) é uma doença autossômica recessiva, que apresenta 34 mutações no gene GLUT2 e cujo defeito enzimático subjacente ainda é desconhecido.
SINTOMAS: hepatomegalia, acidose tubular renal proximal e retardo de crescimento acentuado, com acúmulo de glicogênio no fígado e nos rins. Ao nascimento pode manifestar-se com diabetes neonatal ou cetoacidose diabética.
TRATAMENTO: Restrição da ingestão de galactose, suplementação de água, amido de milho, eletrólitos e vitamina D.
A glicogenose tipo XII está associada a um quadro de miopatia e anemia hemolítica hereditária, causada pela substituição de um único aminoácido dentro da estrutura tetrâmica da enzima aldolase A, que também está presente no músculo esquelético e eritrócitos.
SINTOMAS: intolerância ao exercício e fraqueza após doença febril. Pode ser observada rabdomiólise com elevações da creatina quinase, aspartato aminotransferase, alanina aminotransferase, lactato desidrogenase e bilirrubina; diminuição na concentração de hemoglobina e hematócrito.
DIAGNÓSTICO: através de testes bioquímicos e hematológicos.
A glicogenose tipo 0 é caracterizada pela deficiência na enzima glicogênio sintase, que afeta primordialmente o fígado. As principais manifestações são hipoglicemia e hipercetonomia ao jejum, e após as refeições apresentam hiperglicemia, hiperlactacidemia e hiperlipidemia.
TRATAMENTO: baseia-se em uma dieta hiperproteica e com carboidratos complexos de baixo índice glicêmico. É indicado o uso noturno de amido de milho cru.
Fonte: Glicogenoses hepáticas: estudo do uso de diferentes amidos e caracterização do perfil de parâmetros do metabolismo do ferro.
Tatiéle Nalin. Tese de doutorado. Nov 2015
Fonte: Translational Science of Rare Diseases 1 (2016) 45–72. DOI 10.3233/TRD-160006
Fonte: M.M. Adeva-Andany et al. / BBA Clínical 5 (2016) 85–100
Fonte: 10.1097/MCO.0000000000000181
Diagnóstico
- Sinais e Sintomas
Variam de acordo com o tipo de glicogenose.
Na GSD 1A os sintomas podem estar presentes ao nascimento ou nas primeiras semanas de vida. O mais comum é o aparecimento, aos 3 - 4 meses de vida, de hepatomegalia ou convulsão associada à hipoglicemia. Alterações frequentemente descritas incluem face de boneca, obesidade troncular, abdômen globoso pela hepatomegalia, resultante do acúmulo de glicogênio e esteatose hepática, hipotonia e baixa estatura. Também pode ocorrer anemia, manifestações intestinais, como diarreia intermitente, enteropatia relacionada à GSD e dano da mucosa intestinal. Associação à neutropenia e/ou infecções recorrentes pode ser observada na GSD Ib.
Os exames podem mostrar hipoglicemia, aumento de lactato, aumento de ácido úrico, hipertrigliceridemia e hiperlipidemia, especialmente após um curto período de permanência em jejum.
Hipoglicemia: muitos bebês apresentam hipoglicemia já nos primeiros dias após o nascimento, após curtos períodos de jejum ou infecção, ou ainda, crianças podem apresentar, como primeiro sintoma, convulsões secundárias à hipoglicemia. Contudo, nem sempre a hipoglicemia é acompanhada de sintomas, pois o cérebro pode fazer uso do ácido láctico como substrato. Com o avançar da idade, e o tratamento correto, há uma tendência à diminuição das hipoglicemias.
Acidose láctica:o lactato pode estar aumentado em cerca de quatro vezes o normal. O ácido láctico é produzido nos músculos e hemácias, é metabolizado no fígado, e desviado para a síntese de ácidos graxos ou gliconeogênese, que, como não ocorre em pacientes com glicogenose tipo I, ocorre seu acúmulo. Com o controle da doença, os níveis podem atingir valores normais.
Aumento do ácido úrico: a hiperuricemia resulta da diminuição da excreção de urato pelo rim (devido competição com ácido láctico) e do aumento na produção de ácido úrico. Como consequência podem aparecer cálculos renais, gota e nefropatia.
Hiperlipidemia: a deposição anormal de gordura pode resultar em algumas características clínicas da doença, como obesidade troncular e "face de boneca”. O acúmulo de gordura pode influenciar ainda no aumento do fígado, juntamente com o acúmulo de glicogênio. Os níveis séricos de triglicerídios podem estar muito elevados, podendo atingir níveis de 4000-6000mg/dL. Pode ocorrer ainda um aumento moderado dos níveis de colesterol e fosfolípides.
Desordens hematológicas: pode ocorrer disfunção das plaquetas (reduzida adesividade e alteração na agregação) levando a um tempo de sangramento prolongado, resultando em queixa de sangramentos nasais e tendência para hemorragia durante procedimentos cirúrgicos. Essas alterações podem ser corrigidas com o controle da doença.
Alteração nos leucócitos polimorfonucleares: O subtipo Ib apresenta neutropenia associada, podendo atingir valores abaixo de 1500/mm. Há ainda uma deficiência na função dessas células, contribuindo para o aumento da susceptibilidade desses pacientes às infecções.
Alterações gastrointestinais: frequentemente pode ocorrer diarreia intermitente, inclusive secundária à sobrecarga de lactose e glicose. Recentemente, há relatos de associação da glicogenose tipo I com doença inflamatória intestinal.
Adenomas e neoplasias hepáticas: os adenomas ocorrem em cerca de 50-70% dos pacientes a partir da segunda e terceira décadas de vida, sendo mais comuns o hepatoma e o adenoma hepatocelular com displasia, que são detectados por meio da ultra-sonografia ou tomografia computadorizada. Com o controle metabólico e o controle dietético, os adenomas podem diminuir de tamanho ou tornar-se indetectáveis, ou ainda, podem sofrer transformação maligna. Os níveis de a-feto-proteína são úteis na monitorização da progressão do tumor.
A GSD também pode manifestar-se com doença renal progressiva, alterações hepáticas teciduais tais como hepatócitos edemaciados, esteatose, hiperglicogenação nuclear e adenomas hepáticos em 22%-75% dos pacientes adultos.
Tipos musculares como a Tipo V podem apresentar a síndrome de intolerância ao exercício muscular, com mialgia, câimbras, fadiga e fraqueza muscular. Pode ocorrer, após a prática de exercício uma elevação da creatina quinase e rabdomiólise com mioglobinúria (urina escura), que pode levar à insuficiência renal aguda. Há casos de manifestação precoce com hipotonia, fraqueza muscular generalizada e insuficiência respiratória progressiva.
- Exames
Glicose
Lactato
Ácido Úrico
Triglicérides
Colesterol
Ultrasson do fígado
O teste do glucagon não é indicado e pode ser perigoso, pois não elevará o nível de glicose e vai elevar significativamente o ácido lático, podendo provocar hipoglicemias graves e crise convulsiva.
O diagnóstico é clínico, baseado nos sintomas, contudo para confirmação do diagnóstico e definição do tipo de Glicogenose, relevante para o planejamento da terapia, torna-se importante a realização do teste genético.
Importante lembrar que esses exames devem ser realizados apenas por profissionais e com a prescrição do seu médico, que é quem fará a avaliação dos resultados.
HIPOGLICEMIA
GLICEMIA NORMAL
A manutenção rigorosa dos níveis sanguíneos de glicose é fundamental para o funcionamento do cérebro, mesmo que este possa se adaptar a níveis mais baixos ou mesmo utilizar corpos cetônicos oriundos da degradação de gorduras. Após a ingestão de alimentos, a glicose pode ser metabolizada em piruvato ou armazenada na forma de glicogênio no fígado ou nos músculos, para ser utilizada conforme a necessidade. Nos períodos de jejum, ocorre a glicogenólise hepática, que é a quebra do glicogênio armazenado para formar a glicose, um mecanismo que recompõe as taxas de glicose de uma forma rápida. O fígado possui uma importante função na regulação do estoque de glicose e de armazenamento desta. Os valores de referência de normalidade da glicemia são entre 70 mg/dL e 100 mg/dL.
COMO OCORRE A HIPOGLICEMIA
O glicogênio está presente em todas as células das pessoas, mais abundantemente no fígado e nos músculos, sendo a forma através da qual armazenamos a glicose vinda da alimentação. Quando há necessidade do organismo utilizar a glicose (como em situações de stress, jejum), enzimas quebram o glicogênio e liberam a glicose para a circulação sanguínea e, desse modo, para vários órgãos, incluindo o cérebro. Quando o glicogênio não consegue ser quebrado devido à deficiência de algumas das enzimas envolvidas, este se acumula no órgão e, a não liberação de glicose para a circulação acarreta em hipoglicemia e uma série de consequências.
SINTOMAS DA HIPOGLICEMIA
No corpo: desmaio, fadiga, fome, fome excessiva, sudorese excessiva, tontura e tremores.
Na cognição: apatia ou confusão mental.
No humor: ansiedade ou nervosismo.
No coração: palpitações ou ritmo cardíaco acelerado.
Na boca: formigamento nos lábios ou secura.
Também pode se manifestar com: dor de cabeça, fala arrastada, irritabilidade, palidez, pupila dilatada, sensação de formigamento, sonolência, tremor ou visão embaçada.
DESCOMPENSAÇÃO
Se você detectar um valor muito baixo de glicose no sangue, deve procurar um médico ou um serviço de saúde o mais rápido possível, para que seja realizado o diagnóstico e eventual tratamento.
Coma hipoglicêmico ocorre, geralmente, em pacientes que ficam sem tratamento até que a glicemia reduza ao ponto de causar alterações do metabolismo, comprometendo a consciência. Nesse caso, a pessoa deve ser encaminhada, imediatamente, a um serviço de emergência médica.
O paciente pode evoluir para acidose metabólica, necessitando correção imediata.
TRATAMENTO
O tratamento das glicogenoses hepáticas consiste na administração frequente de amido de milho cru (para glicogenoses hepáticas), visando à manutenção da normoglicemia (> 75 mg/dL) e a prevenção de distúrbios metabólicos secundários.
A administração do amido de milho cru deve ser seguida conforme a prescrição e orientação do médico de acordo com cada tipo de glicogenose. A média para o cálculo de crianças menores de dois anos de idade é de 1.6g/Kg de peso da criança (a cada 3-4 horas), e para crianças maiores de dois anos de idade é de 1.75 a 2.5 g/Kg de peso da criança (a cada 3-4 horas). O tratamento é bem-individualizado e varia de acordo com a resposta de cada paciente. Crianças menores podem não conseguir manter a glicemia com esse intervalo de tempo, devendo ter a sua prescrição ajustada.
O amido de milho cru deve ser pesado em balança para um correto tratamento, evitando o excesso de ingestão de amido e obesidade desnecessária, e também evitando o tratamento insuficiente, não mantendo a glicemia em níveis normais. A ingestão do amido deve ser exatamente nos horários prescritos pelo médico. A medição da glicemia capilar é necessária por várias vezes ao dia para monitorar a glicemia.
Restrições alimentares e suplementações dietéticas são realizadas de acordo com os diferentes tipos de glicogenose também. O paciente deve ser criteriosamente monitorado quanto à reposição de polivitamínicos com vitaminas do complexo B1 e B12, vitamina D, vitamina C e Cálcio.
Para GSD Ib, acrescenta-se ainda o uso de estimulador de colônia de neutrófilos, na dose diária recomendada de 1-2.5 ug/Kg/dia.
A adesão ao tratamento é um grande desafio tanto para pacientes como seus familiares ou cuidadores, exigindo cuidados rigorosos e muita disciplina.
IMPORTÂNCIA DE CONHECER O TIPO
Cada tipo de glicogenose possui características e tratamentos diferentes. É de extrema importância conhecer o tipo específico para não realizar o tratamento de forma errada e agravar o quadro de saúde.
IMPORTÂNCIA DO CONTROLE DA GLICOGENOSE
Fazer o controle da Glicogenose de forma adequada irá trazer mais qualidade de vida e evitar complicações como baixa estatura, hepatomegalia, nefropatias, nódulos hepáticos, hipoglicemias severas e suas consequências, sequelas neurológicas, convulsões, coma e até o óbito.
Conheça a doença e assim poderá desenvolver formas de realizar um controle efetivo e com bons resultados. Com o cuidado adequado é possível ter uma vida com desenvolvimento normal e resultados metabólicos dentro das faixas de normalidade.
CUIDANDO DO TODO
Receber o diagnóstico de uma doença crônica ou mesmo lidar com o dia a dia da doença não é uma tarefa fácil. Não se esqueça que tão importante quanto fazer o monitoramento constante da glicemia e a restrição dos açúcares é o cuidado com o turbilhão de emoções que acompanham esse desafio.
Busque sempre o apoio da família e dos amigos, troque experiências e não exite em procurar ajuda de profissionais caso seja necessário. A estabilidade emocional é peça fundamental para os bons resultados do controle da doença.
ALIMENTAÇÃO
A alimentação correta é a chave do sucesso no controle da Glicogenose. É preciso ter disciplina tanto no uso regular do amido de milho cru quanto na restrição dos alimentos de acordo com cada tipo de glicogenose.
As restrições alimentares variam de acordo com o tipo de glicogenose. Siga rigorosamente a dieta conforme o seu tipo, pois esse ato pode ser primordial para o adequado controle das glicemias e da doença, impactando de forma direta no bom desenvolvimento do organismo e evitando agravos aos pacientes. Há restrição de alimentos que possuam SACAROSE E LACTOSE e limitação de FRUTOSE, a fim de evitar o depósito de glicogênio no fígado. À exceção do tipo III, que não possui restrição à lactose.
Busque a orientação do Nutricionista.
Não deixe de conferir nas embalagens dos alimentos a presença de lactose, sacarose e frutose, bem como as quantidades de carboidratos, proteínas, gorduras e sal. Alimentos com a descrição SEM LACTOSE não necessariamente estão isentos dos produtos da lactose, que fazem igualmente mal aos portadores de Glicogenose. As bebidas lácteas que contêm essa afirmação são úteis aos intolerantes à lactose, pois vem associado à enzima lactase, o que não faz o alimento ser permitido para quem tem Glicogenose.
. CARBOIDRATOS: Considerando que o amido de milho, tão necessário para o controle da glicemia dos pacientes com glicogenose, é carboidrato e de uso obrigatório e em grandes quantidades ao longo do dia, outros carboidratos devem ser consumidos em menores quantidades durante as refeições. O consumo deve ser com moderação e inclui pães, massas, arroz branco, batata, mandioca, iogurtes etc.
. PROTEINAS E GORDURAS: São fundamentais para garantir uma alimentação equilibrada e favorecer o equilíbrio e o crescimento do portador de glicogenose. Dar preferência para azeite de oliva e carnes magras.
. FIBRAS: As fibras estão presentes nos pães integrais, feijão, arroz integral, cevada, aveia, frutas e vegetais frescos.
EXERCÍCIO FÍSICO: Além dos benefícios à saúde, são importantes para ajudar no controle de peso e da glicemia.
Defina com o seu médico e nutricionista como será a alimentação para a prática de exercícios físicos. Lembre-se de tomar o seu amido de milho nos horários indicados e levar consigo algum alimento que eleve a glicemia rapidamente, caso ocorra uma queda brusca dela durante os exercícios.
Leve sempre consigo alguma identificação que revele que possui glicogenose, pois caso tenha algum mal-estar e precise de atendimento médico, facilitará as providências adequadas.
O controle da glicemia deve ser realizado para evitar a ocorrência de hipoglicemia.
HERANÇA GENÉTICA
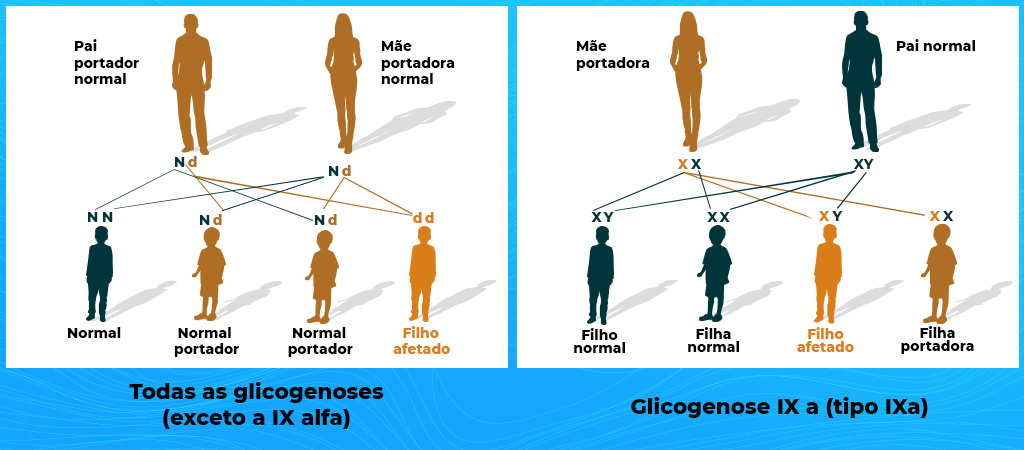
Todas as glicogenoses hepáticas (exceção da IX alfa) possuem um padrão de herança autossômico recessivo, com risco de recorrência de 25% para as próximas gerações, independentemente do sexo da criança ou de já ter outros filhos que tenham a doença. A transferência do gene para os filhos independe também dos pais serem parentes. Pais não parentes podem ter o gene afetado e passarem aos filhos. O grau de parentesco interfere na chance dos pais terem o gene afetado, pois como a doença é genética, parentes tem mais chance de terem uma herança genética semelhante, aumentando a chance de receberem o gene afetado pela glicogenose dos familiares.
Essa recorrência de 25% ocorre quando pai e mãe possuem um gene afetado e um gene normal, sendo ambos portadores do gene afetado, mas não possuindo a doença. Isso acontece pois como a doença é recessiva, ela só se desenvolve quando a pessoa possui os dois genes afetados. Ao passar um gene para o filho/filha, a combinação de genes pode resultar em 25% de chance de ser normal (recebeu dois genes normais), 50% de chance de ser portador de um gene alterado e um gene normal (sendo então portador do gene, mas sem possuir a doença), e em 25% dos casos, receberá o gene afetado tanto do pai quanto da mãe, possuindo os dois genes afetados, e nesse caso, possuindo a Glicogenose.
A chance de uma pessoa com glicogenose ter um filho/filha também com glicogenose vai depender se o outro genitor possui ou não o gene da glicogenose. Se o outro genitor também possuir glicogenose, a recorrência é de 100% do filho/filha ter glicogenose. Se o outro genitor não possuir nenhum gene afetado, todos os filhos receberão um gene afetado e um normal, sendo portadores de um gene, mas sem desenvolver a doença. Mas se o outro genitor possuir um gene afetado e um normal, a chance é de 50% receberem um gene normal e um afetado, sendo portadores somente, e 50% de receber os dois genes afetados, possuindo glicogenose.
A exceção ocorre com a glicogenose tipo IX, que possui um padrão de herança ligado ao cromossomo X, em que a mãe portadora passa o cromossomo X afetado para o filho. Nesse caso, se for menina, ela tem 50% de chance de não receber o gene afetado e ser normal e 50% de chance de receber o X afetado da mãe e ser também portadora. Já se for menino, ele tem 50% de chance de ser normal, e 50% de chance de receber o X afetado da mãe, e possuir glicogenose.
Associe-se
Para fazer parte da Associação Brasileira de Glicogenose - ABGlico preencha a ficha cadastral abaixo. Aguarde que entraremos em contato com você.
Fale conosco
Entre em contato com a ABGlico!
Ficaremos felizes em responder às suas dúvidas e em aceitar sugestões.
Para tanto, preencha o formulário
ao lado e mande pra gente!